Cancer is one of the most complex and instructive human diseases, representing a dramatic disruption of normal cellular processes that allows cells to grow uncontrollably, resist death, invade surrounding tissues, and metastasize. With the incidence of cancer rising worldwide, understanding its underlying mechanisms is critical for students, researchers, and clinicians. Decades of research have revealed that cancer can be understood through distinct but interconnected conceptual dimensions, including:
Acquired functional capabilities (hallmarks of cancer)
Enabling phenotypic characteristics
Hallmark-conveying cells in the tumor microenvironment (TME)
Systemic interactions within the host
Collectively, these dimensions provide a framework to study cancer as a dynamic, adaptive “outlaw organ”, guiding the development of therapeutic strategies.
Introduction: The Hallmarks Concept
The concept of “hallmarks of cancer” was introduced by Douglas Hanahan and Bob Weinberg to organize the vast diversity of genetic and phenotypic changes observed in tumors. The goal was to understand how cancers arise through multistep tumorigenesis, evolve through selective pressures, and acquire traits such as metastasis, therapy resistance, and immune evasion.
Timeline of hallmark evolution:
2000: Six hallmarks: sustaining proliferative signaling, evading growth suppressors, resisting cell death, replicative immortality, angiogenesis, invasion/metastasis.
2011: Added deregulated metabolism and immune evasion.
2022: Phenotypic plasticity added, emphasizing dynamic adaptation to therapy and environment.
A central realization was that mutant cancer cells alone do not define tumor biology. Instead, cancer progression depends on recruitment and reprogramming of surrounding normal cells, creating a supportive tumor microenvironment (TME).
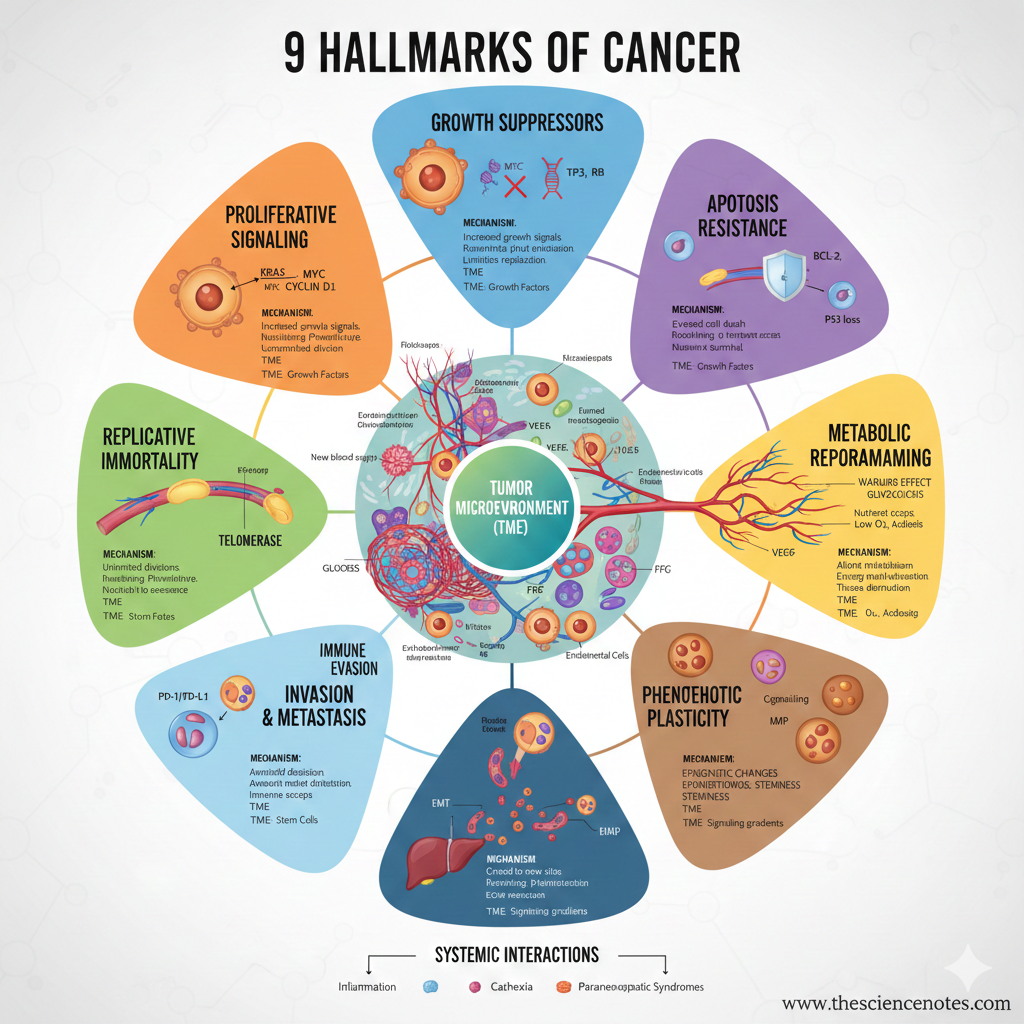
The 9 Hallmarks of Cancer
1. Sustaining Proliferative Signaling
Cancer cells achieve uncontrolled proliferation by activating oncogenes, which drive chronic cell cycle progression.
Key oncogenes:
KRAS, NRAS, HRAS: Mutations in KRAS are seen in ~30% of tumors, including pancreatic (~90%), colorectal (~50%), and lung (~35%) cancers.
BRAF, PIK3CA, BCR-ABL: Drive aberrant growth signals.
MYC: Transcription factor regulating thousands of genes; amplified in ~40% of tumors.
Mechanisms:
Gene amplification or rearrangement
Circular extrachromosomal DNA (ecDNA) enhancing oncogene expression
Epigenetic reprogramming, including autocrine/paracrine growth factor loops
Clinical relevance: Targeted therapies include BRAF inhibitors (vemurafenib) and HER2 inhibitors (trastuzumab).
Additional insight: RAS and MYC can stimulate multiple other hallmark capabilities, including metabolic reprogramming and angiogenesis, highlighting their centrality in tumor evolution.
2. Evading Growth Suppressors
Normal cells regulate proliferation via tumor suppressor genes (TSGs), which act as gatekeepers for cell cycle checkpoints.
Key TSGs:
TP53: Activated in response to DNA damage, oncogenic stress, or hypoxia; regulates apoptosis, senescence, and cell-cycle arrest. Mutated in ~40% of cancers.
RB, CDKN1A/B (p21/p27), CDKN2A (p16INK4a/p14ARF): Block progression through G1/S and G2/M transitions.
APC: Degrades β-catenin to prevent uncontrolled proliferation.
Mechanisms of evasion:
Mutations or deletions
Epigenetic silencing
Circumventing paracrine growth suppression (e.g., contact inhibition or competition for nutrients)
Clinical relevance: Drugs like CDK inhibitors (palbociclib, ribociclib) can restore growth suppression in tumors with TSG inactivation.
3. Resisting Programmed Cell Death (Apoptosis)
Cancer cells evade cellular suicide mechanisms, enabling survival despite DNA damage or abnormal signaling.
Mechanisms:
Overexpression of anti-apoptotic proteins: BCL-2, BCL-XL, MCL-1
TP53 inactivation, preventing induction of pro-apoptotic genes like PUMA and NOXA
Dysregulation of alternative cell death pathways: necroptosis, ferroptosis, pyroptosis, autophagy
Therapeutics: BH3 mimetics (Venetoclax) restore apoptosis in CLL and AML, with ongoing trials in other cancers.
Paradoxical insight: Apoptotic cells can stimulate tumor-promoting signals in neighboring cells or escape death with partially damaged genomes, contributing to genomic instability and tumor progression.
4. Establishing Replicative Immortality
Normal cells are limited by a mitotic clock dictated by telomere length. Cancer cells bypass this to divide indefinitely.
Mechanisms:
Telomerase activation (TERT): Adds telomere repeats, common in glioblastoma (~80%), melanoma (~60%), bladder cancer (~80%).
Alternative Lengthening of Telomeres (ALT): Recombination-based telomere extension, common in mesenchymal and neuroepithelial tumors.
Consequences:
Chromosomal instability facilitates acquisition of other hallmark traits.
Delayed telomerase activation allows mutational selection and tumor evolution.
5. Inducing or Accessing Vasculature (Angiogenesis)
Tumors require oxygen and nutrients to grow beyond 1–2 mm.
Mechanisms:
Hypoxia triggers VEGFA, ANGPT2, and FGF secretion
Endothelial cell activation, sprouting, and capillary formation
Recruitment of pericytes for vessel stabilization
Tumor vasculature:
Leaky, chaotic, and poorly perfused
Impedes immune cell infiltration, contributing to immune evasion
Alternative mechanism: Vascular co-option, where tumors hijack pre-existing vessels, particularly after anti-angiogenic therapy.
Clinical relevance: Anti-angiogenic therapy (e.g., bevacizumab) targets VEGF pathways.
6. Deregulating Cellular Metabolism
Cancer cells reprogram metabolism to meet energy and biosynthetic demands.
Metabolic strategies:
Aerobic glycolysis (Warburg effect) alongside oxidative phosphorylation
Utilization of alternative fuels: lactate, glutamine
Metabolic crosstalk with tumor microenvironment cells (fibroblasts, macrophages, T cells)
TME factors:
Hypoxia, acidosis, nutrient gradients
Paracrine secretion of metabolites
Dynamic adaptations during tumor progression and metastasis
Therapeutic implications: Targeting glycolysis or glutamine metabolism can disrupt tumor growth.
7. Activating Invasion and Metastasis
Cancer cells acquire the ability to spread beyond their origin.
Mechanisms:
Epithelial-to-mesenchymal transition (EMT) → motility
Extracellular matrix remodeling via MMPs
Entry into blood or lymphatic circulation
Colonization of distant organs
TME contribution: Stromal cells and immune cells secrete factors facilitating invasion.
Clinical importance: Metastasis causes ~90% of cancer-related deaths.
8. Evading Immune Destruction
Tumors escape immune surveillance via:
Immune checkpoints (PD-L1, CTLA-4)
Immunosuppressive cytokines
Remodeling of tumor vasculature to prevent T-cell infiltration
Therapeutics: Checkpoint inhibitors (nivolumab, pembrolizumab) restore T-cell activity.
9. Unlocking Phenotypic Plasticity
Cancer cells dynamically switch between proliferative, invasive, and drug-resistant states.
Significance:
Drives therapy resistance, relapse, and metastasis
Allows adaptation to fluctuating TME and systemic pressures
Enabling Phenotypic Characteristics
These support the acquisition of hallmarks:
Genomic instability: accelerates mutations and tumor evolution
Tumor-promoting inflammation: certain immune cells facilitate growth
Epigenetic remodeling and oxidative stress adaptation enhance survival under adverse conditions
Tumor Microenvironment (TME)
Tumors are heterogeneous “organs” with multiple interacting cell types:
Cancer cells: proliferate and adapt
Fibroblasts: remodel ECM and secrete growth factors
Immune cells: suppress or promote tumor growth
Blood vessels: deliver nutrients and remove waste
Role: The TME contributes to growth, invasion, angiogenesis, immune evasion, metabolic adaptation, and therapy resistance.
Systemic Interactions
Tumors interact with the body systemically, altering:
Hormone levels
Metabolic balance
Immune surveillance
Impact: Systemic effects influence tumor progression and therapy response.
Therapeutic Implications
Effective cancer therapy often requires multi-hallmark targeting:
Oncogene inhibitors: KRAS, BRAF, HER2
Tumor suppressor modulators: CDK inhibitors
Apoptosis activators: BH3 mimetics
Anti-angiogenic drugs: VEGF inhibitors
Immunotherapy: checkpoint inhibitors
Metabolic inhibitors: glycolysis/glutamine targeting
Combination therapy is crucial to overcome plasticity, adaptation, and resistance.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hanahan, D. (2022). Hallmarks of cancer: New dimensions. Cancer Discovery, 12(1), 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059
Hanahan, D. (2026). Hallmarks of cancer—Then and now, and beyond. Cell. https://doi.org/10.1016/j.cell.2025.12.049