Introduction to Hi-C Technology
Hi-C TECHNOLOGY is a powerful genome-wide chromosome conformation capture technique used to identify long-range chromatin interactions in an unbiased and high-throughput manner. By combining proximity ligation with next-generation sequencing, Hi-C enables researchers to explore the three-dimensional (3D) organization of the genome and understand how chromatin folding influences gene regulation, genome stability, and cellular identity.
Since its development, Hi-C has transformed our understanding of nuclear architecture, revealing fundamental principles such as chromosome territories, A/B compartmentalization, and fractal globule chromatin organization.
Principle of the Hi-C Method
The Hi-C technique is based on a simple but elegant idea: DNA segments that are close together in the nucleus are more likely to be ligated together after cross-linking. By capturing and sequencing these ligation products, Hi-C generates a genome-wide map of chromatin contacts.
Key features of Hi-C include:
Genome-wide and unbiased detection of interactions
Detection of both intra-chromosomal and inter-chromosomal contacts
Applicability across multiple scales, from kilobases to entire chromosomes
Step-by-Step Overview of the Hi-C Experimental Workflow
1. Cross-Linking and Cell Lysis
Hi-C begins with formaldehyde cross-linking of cells to preserve native chromatin interactions. Typically, 2 × 10⁷ to 2.5 × 10⁷ mammalian cells are used. Cells are lysed, and nuclei are isolated to release chromatin while maintaining cross-linked DNA–protein complexes.
2. Restriction Enzyme Digestion
The cross-linked chromatin is digested using a restriction enzyme (commonly HindIII), fragmenting the genome at specific recognition sites. This step generates sticky ends that reflect the linear genome organization prior to ligation.
3. Biotin Fill-In and End Labeling
The digested DNA ends are filled in to create blunt ends, incorporating biotinylated dCTP in the process. This biotin label marks DNA ends that will later participate in ligation junctions, enabling selective purification.
A parallel sample without biotin labeling is retained as a 3C control to assess digestion and ligation efficiency.
4. Proximity Ligation Under Dilute Conditions
To favor ligation between spatially proximal, cross-linked DNA fragments, ligation is performed under extremely dilute conditions using T4 DNA ligase. This step creates chimeric DNA molecules that represent physical chromatin interactions inside the nucleus.
5. Cross-Link Reversal and DNA Purification
Proteinase K treatment reverses cross-links and removes proteins. The DNA is then purified through phenol–chloroform extraction and ethanol precipitation, yielding high-molecular-weight ligation products.
High-quality Hi-C libraries appear as tight bands larger than 10 kb, while smearing indicates poor ligation efficiency.
Library Preparation for High-Throughput Sequencing
6. DNA Shearing and End Repair
Purified Hi-C DNA is sheared to 300–500 base pairs using acoustic sonication. Fragmented DNA undergoes end repair and A-tailing to prepare it for adapter ligation.
7. Biotin Pull-Down of Ligation Junctions
Biotinylated ligation junctions are selectively enriched using streptavidin magnetic beads. This step ensures that only DNA fragments representing true chromatin interactions are sequenced.
8. Adapter Ligation and PCR Amplification
Illumina paired-end sequencing adapters are ligated directly on bead-bound DNA. The library is then PCR-amplified using an optimized number of cycles, determined by test reactions to avoid over-amplification.
The final Hi-C library is purified and validated before sequencing.
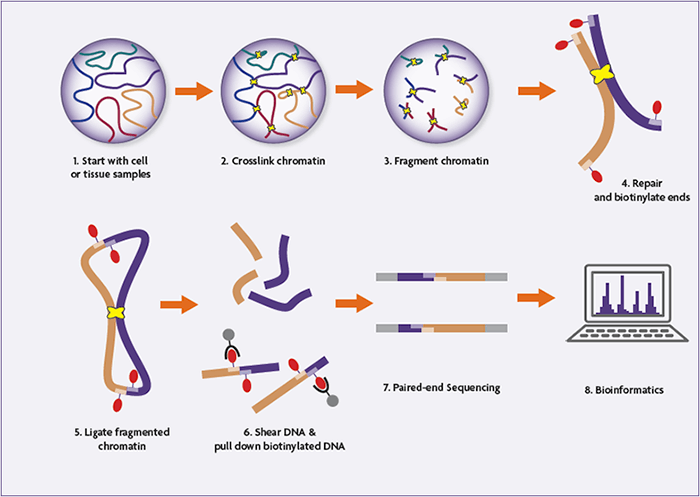
Hi-C Sequencing and Data Analysis
Hi-C libraries are sequenced using paired-end next-generation sequencing, and each read end is aligned independently to the reference genome. Interacting fragment pairs are then reconstructed computationally.
Expected Interaction Distributions
In a successful Hi-C experiment:
~55% of read pairs represent inter-chromosomal interactions
~15% are short-range intra-chromosomal interactions (<20 kb)
~30% are long-range intra-chromosomal interactions (>20 kb)
These metrics serve as important quality control benchmarks.
Visualization of Chromatin Interactions
Hi-C Heat Maps
Chromatin interactions are commonly visualized as heat maps, where:
The x- and y-axes represent genomic coordinates
Each pixel represents interaction frequency
A strong diagonal reflects frequent interactions between nearby genomic loci.
Chromosome Territories
Hi-C data show that intra-chromosomal interactions are enriched compared to inter-chromosomal interactions, providing direct evidence for chromosome territories—a foundational principle of nuclear organization.
A/B Compartmentalization of the Genome
Correlation analysis of Hi-C contact matrices reveals that the human genome segregates into two major compartments:
A compartment: gene-rich, transcriptionally active, open chromatin
B compartment: gene-poor, transcriptionally inactive, closed chromatin
This segregation appears as a characteristic plaid pattern in correlation heat maps.
Fractal Globule Model of Chromatin Folding
One of the most significant insights from Hi-C was evidence supporting the fractal globule model of chromatin organization.
Key observations include:
Contact probability scales with genomic distance following a power law with a slope of approximately −1
This behavior is inconsistent with an equilibrium globule but matches predictions for a fractal globule
Why Fractal Globules Matter
They are knot-free and unentangled
Genomic regions close along the linear genome remain close in 3D space
They can rapidly unfold and refold, supporting dynamic gene regulation
Resolution and Sequencing Depth
The resolution of Hi-C interaction maps depends directly on sequencing depth:
~30 million reads yield ~1 megabase resolution
Increasing resolution by a factor of n requires n² more reads
This quadratic relationship is critical when designing Hi-C experiments.
Applications of Hi-C
Hi-C is widely used to:
Study genome architecture and nuclear organization
Identify regulatory interactions between enhancers and promoters
Understand chromatin changes in development and disease
Improve genome assemblies and structural variant detection
Conclusion
The Hi-C method enables unbiased, genome-wide identification of chromatin interactions and provides unprecedented insights into the 3D structure of the genome. From chromosome territories and compartmentalization to fractal globule organization, Hi-C has reshaped how we understand genome function beyond linear DNA sequence.
With proper controls, sufficient sequencing depth, and rigorous data analysis, Hi-C remains a cornerstone technology for modern genomics and epigenetics research.