What is urea cycle?
The urea cycle is a type of metabolism in the liver that that changes toxic ammonia into less toxic urea for healthy removal, which is necessary for the body’s nitrogen balance.
Steps of Urea cycle
Step 1:
Formation of Carbamoyl Phosphate
The cycle begins with ammonia and carbon dioxide condensation, which is mediated by the enzyme carbamoyl phosphate synthetase I (CPS I). This process produces carbamoyl phosphate, which is then employed in the urea cycle.
Step 2:
Formation of Citrulline
The second stage includes the condensation of carbamoyl phosphate with ornithine, which is mediated by the enzyme ornithine transcarbamylase (OTC). This leads in the production of citrulline.
Step 3:
Formation of Arginosuccinate
The enzyme argininosuccinate synthetase (ASS) catalyzes the addition of aspartate to citrulline in the third stage. As a result, argininosuccinate is formed.
Step 4:
Formation of Arginine and Fumarate
In the fourth phase, argininosuccinate is cleaved by argininosuccinate lyase (ASL) to generate arginine and fumarate. The fumarate is utilized in the citric acid cycle, whereas arginine is used in the urea metabolism.
Step 5:
Formation of Urea
Finally, arginase hydrolyzes arginine to produce urea and ornithine. Urine excretes urea, whereas ornithine is reused in the cycle.
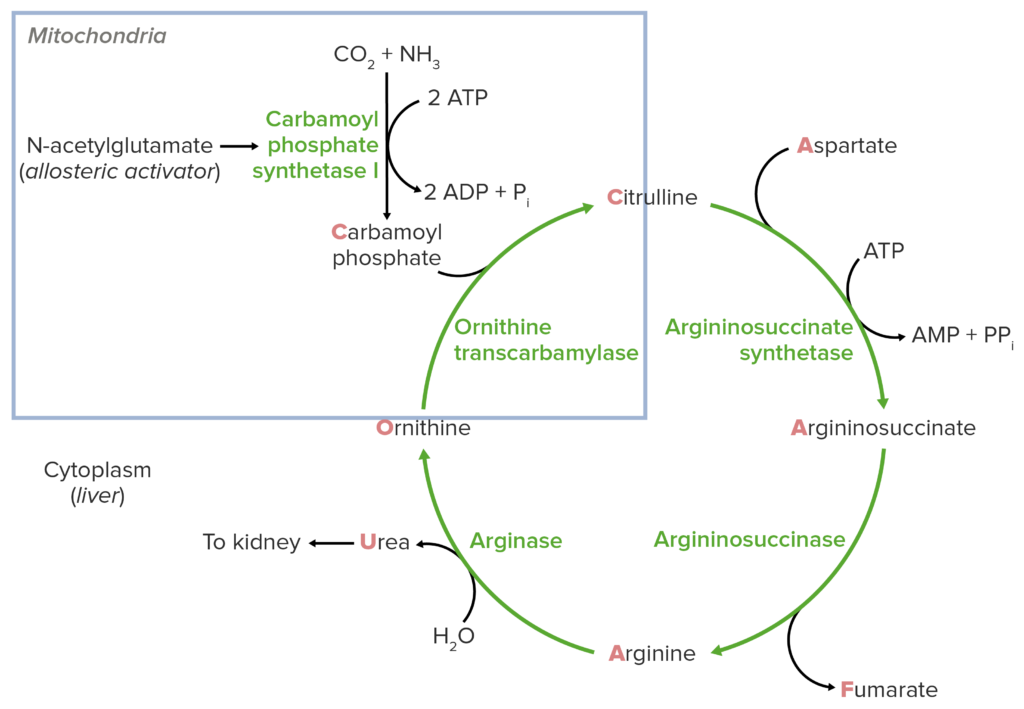
The urea cycle biochemical processes occur in both the mitochondrial and cytosolic compartments of liver cells.
Mitochondrial Stage
The first two stages of the urea metabolism take place in the mitochondrial stage. CPS I is an enzyme that catalyzes the conversion of ammonia and bicarbonate to carbamoyl phosphate utilizing ATP as a source of energy. This process occurs in the mitochondrial matrix, which contains CPS I. The carbamoyl group is transferred from carbamoyl phosphate to ornithine in the second step of the mitochondrial stage, resulting in citrulline. The enzyme ornithine transcarbamylase (OTC), which is also found in the mitochondrial matrix, catalyzes this process.
Cytosolic stage
After citrulline gets produced, it travels from the mitochondria to the cytosol, where the rest of the urea metabolic cycle occurs. This is referred to as the urea cycle’s cytosolic step. Citrulline interacts with aspartate in the cytosol to create argininosuccinate, which is mediated by the enzyme argininosuccinate synthetase (ASS). The enzyme argininosuccinate lyase (ASL) then cleaves argininosuccinate into fumarate and arginine.
The urea metabolic cycle concludes with the change of arginine to urea and ornithine, which is mediated by the enzyme arginase. Urea is eliminated in the urine, whereas ornithine is carried back to the mitochondria to restart the cycle.
Regulation of Urea cycle
The urea cycle is regulated at several levels, including transcriptional, translational, and post-translational control.
- N-acetylglutamate (NAG) is an important urea metabolism regulator because it stimulates the first enzyme in the route, carbamoyl phosphate synthetase I (CPS I).
- The availability of substrates such as ammonia and bicarbonate can influence the urea metabolism rate.
- Transcription factors such as CCAAT/enhancer-binding protein (C/EBP) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1) influence the expression of genes involved in the cycle.
- Feedback inhibition governs the cycle as well, with high arginine levels blocking the enzyme argininosuccinate synthetase (ASS).
- Hormones like as glucagon and insulin can also influence the cycle by influencing the supply of substrate and gene expression.
Urea cycle disorders
Dysregulation of the urea cycle can result in hyperammonemia, which is an abnormal buildup of ammonia in the blood. This can result in a variety of health issues, including:
- Neurological damage: High amounts of ammonia can induce brain and nervous system damage, resulting in symptoms including disorientation, convulsions, and coma.
- Liver damage: Because the liver is important for converting ammonia to urea, urea metabolism dysregulation can potentially cause liver damage.
- Delays in development: Hyperammonemia can cause developmental delays in newborns and children, especially in cognitive and motor abilities.
- Behavioral changes: Hyperammonemia can produce behavioral and emotional changes such as irritability, aggressiveness, and sadness.
- Breathing problems: Hyperammonemia can induce respiratory discomfort and breathing difficulty in severe situations.
Dysregulation of the urea metabolism can be caused by a range of genetic and environmental factors, including:
- Genetic mutations: Inherited genetic mutations can disrupt the enzymes involved in the urea metabolism, resulting in dysregulation and hyperammonemia.
- Liver disease: Liver disease can limit the liver’s capacity to process ammonia, resulting in hyperammonemia.
- Medications: Certain drugs, such as valproic acid, might disrupt the urea metabolism and produce hyperammonemia.
- Nutritional deficiencies: Nutrient shortages such as Vitamin B12, folic acid, and biotin can disrupt the action of enzymes involved in the urea cycle, resulting in dysregulation.
Treatment for hyperammonemia include addressing the underlying cause and lowering blood ammonia levels. This may include medicines, dietary modifications, and other therapies to promote liver function and eliminate excess ammonia from the body. Early identification and treatment of hyperammonemia are critical for avoiding long-term health consequences.
Learn more