What is Electrophoretic Mobility Shift Assay (EMSA)?
Electrophoretic Mobility Shift Assay (EMSA) is a biochemical method used to examine how proteins interact with nucleic acids like DNA or RNA as well as DNA-RNA interactions. It is also known as a band shift assay or a gel shift test.
In this assay, a combination of proteins and nucleic acids is electrophoresed on a gel. To enable their detection following gel electrophoresis, the nucleic acid fragments are marked with a radioactive or fluorescent tag.
The EMSA can be used to identify the DNA-binding proteins, to determine the specificity and affinity of the protein-DNA interaction, to study the effect of mutations on the interaction, and to identify the binding site of the protein on the nucleic acid sequence.
Principle of Electrophoretic Mobility Shift Assay (EMSA)
EMSA is based on the principle that when the single DNA fragments or double stranded oligonucleotides migrate over nondenaturing polyacrylamide gel, DNA and protein complexes migrate more slowly.
Additionally, another fundamental principle that underpins the technique is the kinetic analysis of EMSA. When a protein (P) binds to a specific DNA (D) site, a complex (PD) is created in equilibrium with the free components:
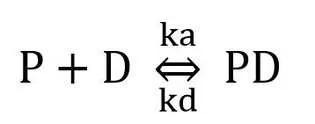
where ka is the association rate and kd is the dissociation rate. A distinct band (PD) is seen when there is a significant contact between protein and DNA, Ka>Kd.
The two main bands would, however, also display a faint smear because electrophoresis causes the separation of the two proteins. Multiple complexes will form and we may see numerous bands if a single DNA molecule has multiple binding sites for a certain protein.
Procedure of Electrophoretic Mobility Shift Assay (EMSA)

- Preparation of the DNA probe: A fluorescent, radioactive, or chemiluminescent tag is added to a short, particular DNA fragment to make it easier to identify it following electrophoresis.
- Preparation of the protein extract: The protein extract is prepared from cells or tissues, and the protein concentration is measured.
- Incubation of the DNA probe with the protein extract: The labeled DNA probe is incubated with the protein extract to allow the formation of protein-DNA complexes.
- Separation of the protein: DNA complexes from unbound DNA: The protein-DNA complexes are separated from unbound DNA by electrophoresis on a polyacrylamide gel.
- Visualization of the protein-DNA complexes: The labeled DNA fragments are visualized using autoradiography, fluorescence or chemiluminescence, and the presence of shifted bands (protein-DNA complexes) is detected.
- Characterization of the protein-DNA complexes: The binding specificity, affinity, stoichiometry, and stability of the protein-DNA complexes can be determined by EMSA by using specific competitors, mutants or antibodies.
General protocol of EMSA
Here is a general protocol for Electrophoretic Mobility Shift Assay (EMSA):
Materials:
- Radioactively-labeled DNA probe
- Protein sample
- Binding buffer (e.g., Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 5% glycerol, 1 mM DTT, 0.05% NP-40)
- Polyacrylamide gel (6% or 8%)
- Loading buffer (e.g., 10 mM Tris-HCl pH 7.5, 10 mM EDTA, 20% glycerol, 0.05% bromophenol blue)
- Electrophoresis buffer (e.g., 0.5x Tris-borate-EDTA (TBE))
- Transfer buffer (e.g., 0.5x TBE, 10% methanol)
- Nitrocellulose membrane
- Blocking buffer (e.g., 5% non-fat dry milk in TBS-Tween)
- Anti-protein antibody conjugated to horseradish peroxidase (HRP)
- Chemiluminescent substrate
Protocol:
- Prepare the radioactively-labeled DNA probe by end-labeling with [γ-32P] ATP using T4 polynucleotide kinase. The probe should be purified by gel electrophoresis and quantified by scintillation counting.
- Prepare the protein sample by diluting it in binding buffer to the desired concentration.
- Mix the protein sample with the radioactively-labeled DNA probe and incubate at room temperature for 20-30 minutes to allow the protein-DNA complex to form.
- Prepare a polyacrylamide gel and pre-run it in electrophoresis buffer for 20-30 minutes.
- Mix the protein-DNA complex with loading buffer and load onto the gel.
- Run the gel in electrophoresis buffer at 100-120 volts until the dye front reaches the bottom of the gel.
- Transfer the protein-DNA complex from the gel to a nitrocellulose membrane using transfer buffer and a transfer apparatus.
- Block the nitrocellulose membrane in blocking buffer for 1 hour at room temperature.
- Incubate the membrane with anti-protein antibody conjugated to HRP diluted in blocking buffer for 1 hour at room temperature.
- Wash the membrane with TBS-Tween buffer (e.g., 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20) to remove unbound antibody.
- Develop the membrane using a chemiluminescent substrate and visualize the protein-DNA complex using autoradiography or a chemiluminescence imaging system.
Note: EMSA can also be performed using non-radioactive detection methods, such as using a biotinylated DNA probe and a streptavidin-HRP conjugate instead of a radioactively-labeled probe and an anti-protein antibody-HRP conjugate. The protocol would be similar, but the detection step would be different.
Non-radioactive EMSA protocol
Here is a non-radioactive Electrophoretic Mobility Shift Assay (EMSA) protocol:
Materials:
- DNA probe labeled with biotin at the 5′ end
- Protein sample
- Binding buffer (e.g., Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 5% glycerol, 1 mM DTT, 0.05% NP-40)
- Polyacrylamide gel (6% or 8%)
- Loading buffer (e.g., 10 mM Tris-HCl pH 7.5, 10 mM EDTA, 20% glycerol, 0.05% bromophenol blue)
- Electrophoresis buffer (e.g., 0.5x Tris-borate-EDTA (TBE))
- Transfer buffer (e.g., 0.5x TBE, 10% methanol)
- Nitrocellulose membrane
- Blocking buffer (e.g., 5% non-fat dry milk in TBS-Tween)
- Streptavidin-HRP conjugate
- Chemiluminescent substrate
Protocol:
- Prepare the biotin-labeled DNA probe by annealing the complementary oligonucleotides in annealing buffer (e.g., 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA). The probe should be purified by gel electrophoresis and quantified by UV spectroscopy.
- Prepare the protein sample by diluting it in binding buffer to the desired concentration.
- Mix the protein sample with the biotin-labeled DNA probe and incubate at room temperature for 20-30 minutes to allow the protein-DNA complex to form.
- Prepare a polyacrylamide gel and pre-run it in electrophoresis buffer for 20-30 minutes.
- Mix the protein-DNA complex with loading buffer and load onto the gel.
- Run the gel in electrophoresis buffer at 100-120 volts until the dye front reaches the bottom of the gel.
- Transfer the protein-DNA complex from the gel to a nitrocellulose membrane using transfer buffer and a transfer apparatus.
- Block the nitrocellulose membrane in blocking buffer for 1 hour at room temperature.
- Incubate the membrane with streptavidin-HRP conjugate diluted in blocking buffer for 1 hour at room temperature.
- Wash the membrane with TBS-Tween buffer (e.g., 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20) to remove unbound streptavidin-HRP conjugate.
- Develop the membrane using a chemiluminescent substrate and visualize the protein-DNA complex using autoradiography or a chemiluminescence imaging system.
Note: Non-radioactive EMSA is a safer alternative to radioactive EMSA, but it may be less sensitive and have higher background noise. Optimization of the protocol is important to minimize non-specific binding and maximize signal-to-noise ratio.
Applications of Electrophoretic Mobility Shift Assay (EMSA)
- Identifying DNA-binding proteins: EMSA can be used to identify and characterize DNA-binding proteins in a complex mixture of proteins. The labeled DNA probe is incubated with the protein extract, and the shifted bands (protein-DNA complexes) are detected, allowing the identification of the DNA-binding proteins.
- Mapping protein-DNA binding sites: EMSA can be used to map the binding site(s) of a protein on a DNA fragment. By using mutant or competitor DNA fragments, the specific binding site(s) of the protein can be identified.
- Studying the specificity of protein-DNA interactions: EMSA can be used to study the specificity of protein-DNA interactions by using competitor DNA fragments that compete with the labeled DNA probe for binding to the protein.
- Studying the affinity of protein-DNA interactions: EMSA can be used to study the affinity of protein-DNA interactions by using different concentrations of the protein extract or by using mutants of the protein or the DNA fragment.
- Studying the effect of post-translational modifications on protein-DNA interactions: EMSA can be used to study the effect of post-translational modifications, such as phosphorylation or acetylation, on protein-DNA interactions by using modified or unmodified protein extracts.
- High-throughput screening of compounds that affect protein-DNA interactions: EMSA can be used in high-throughput screening of small molecules or drugs that affect protein-DNA interactions, allowing the identification of potential therapeutics.
Overall, EMSA is a powerful and versatile technique for the study of protein-nucleic acid interactions, and its applications are widespread in various fields of research, including molecular biology, biochemistry, genetics, and pharmacology.
Controls used in EMSA
To ensure the accuracy and reliability of Electrophoretic Mobility Shift Assay (EMSA) results, it is essential to include appropriate controls. Here are some of the commonly used controls in EMSA:
- Negative control: A negative control is used to ensure that the shifted band observed is due to the protein-DNA interaction and not due to non-specific binding. A negative control consists of a labeled DNA probe incubated with a protein extract that does not bind to the probe, such as a control extract from cells or tissues that do not express the protein of interest.
- Positive control: A positive control is used to ensure that the assay conditions are appropriate for the detection of protein-DNA interactions. A positive control consists of a labeled DNA probe incubated with a protein extract that is known to bind to the probe, such as a control extract from cells or tissues that express the protein of interest.
- Specific competitor control: A specific competitor control is used to confirm the specificity of the protein-DNA interaction observed. A specific competitor is a non-labeled DNA fragment that contains the same binding site(s) as the labeled probe, and it competes with the labeled probe for binding to the protein.
- Non-specific competitor control: A non-specific competitor control is used to control for non-specific binding of the protein to the DNA probe. A non-specific competitor is a non-labeled DNA fragment that does not contain the binding site(s) of the labeled probe, and it competes with the labeled probe for binding to the protein.
- Antibody control: An antibody control is used to confirm the identity of the protein-DNA complex observed. An antibody specific to the protein of interest is added to the incubation mixture to see if it interferes with the formation of the protein-DNA complex, resulting in the loss of the shifted band.
By including these controls in the EMSA experiment, researchers can ensure the specificity, accuracy, and reproducibility of their results.
EMSA Troubleshooting
Electrophoretic Mobility Shift Assay (EMSA) is a sensitive technique, and sometimes, unexpected results or difficulties may arise during the experiment. Here are some common issues that researchers may encounter during EMSA and possible solutions:
- Weak or no shifted band: The weak or no shifted band can be due to low protein concentration, low DNA probe concentration, or non-optimal binding conditions. Increase the protein and/or DNA probe concentration, optimize the binding conditions such as buffer composition, pH, temperature, and incubation time, or try a different protein extract.
- Non-specific binding: Non-specific binding can lead to the formation of multiple shifted bands or smearing. The use of appropriate negative and non-specific competitor controls can help to identify and minimize non-specific binding.
- Multiple shifted bands: Multiple shifted bands can be due to the formation of different protein-DNA complexes. The use of specific competitor controls and antibody supershift assays can help to identify the specific complex of interest.
- High background: High background can be due to the use of excessive amounts of labeled DNA probe or protein extract. Reduce the amount of labeled DNA probe or protein extract, or use fresh reagents.
- Inconsistent results: Inconsistent results can be due to variations in the quality and quantity of protein extract or DNA probe. Ensure that the protein extract and DNA probe are of consistent quality and quantity, and use the same batch of reagents throughout the experiment.
- Protein degradation: Protein degradation can lead to the loss of protein activity, resulting in weak or no shifted band. Ensure that the protein extract is kept on ice or at 4°C, avoid repeated freeze-thaw cycles, and use fresh reagents.
- DNA degradation: DNA degradation can lead to the loss of DNA probe activity, resulting in weak or no shifted band. Ensure that the DNA probe is stored at -20°C or -80°C, avoid repeated freeze-thaw cycles, and use fresh reagents.
By troubleshooting these common issues, researchers can optimize the EMSA protocol and obtain accurate and reliable results.
Limitations of EMSA Assay
Here are some of the limitations of EMSA:
- Limited information: EMSA can provide information about the presence or absence of protein-DNA interactions, but it cannot provide information about the specific binding sites or the binding affinity between the protein and DNA.
- False positives and false negatives: EMSA can give false-positive results due to non-specific binding, and false-negative results due to low-affinity binding or partial dissociation of protein-DNA complexes during gel electrophoresis.
- Requirement for purified proteins: EMSA requires highly purified proteins, which can be time-consuming and expensive to obtain, especially for novel or rare proteins.
- Sensitivity limitations: EMSA may not be sensitive enough to detect low levels of protein-DNA interactions in complex mixtures, such as crude cell extracts.
- DNA probe design limitations: The specificity and efficiency of EMSA depend on the design and quality of the DNA probe used. The DNA probe design may not be optimal for certain proteins or binding sites, which can affect the accuracy and reliability of the results.
- Limited throughput: EMSA is a time-consuming and labor-intensive technique, and it is not suitable for high-throughput analysis of large numbers of samples.
Overall, EMSA is a useful technique for studying protein-DNA interactions, but it should be complemented with other methods, such as chromatin immunoprecipitation (ChIP) and DNA Footprinting, to obtain a more comprehensive understanding of protein-DNA interactions.
Alternatives to EMSA
There are several alternatives to Electrophoretic Mobility Shift Assay (EMSA) for studying protein-DNA interactions. Here are some of the commonly used techniques:
- Chromatin Immunoprecipitation (ChIP): ChIP is a technique that allows the identification of protein-DNA interactions in vivo. It involves crosslinking protein-DNA complexes in cells, followed by immunoprecipitation of the protein of interest, and identification of the associated DNA by PCR or sequencing.
- DNA Footprinting: DNA Footprinting is a technique that can identify protein-DNA interactions by detecting changes in DNA accessibility caused by protein binding. It involves incubating purified DNA with the protein of interest, followed by digestion of unprotected DNA, and identification of the protected DNA regions by gel electrophoresis.
- Surface Plasmon Resonance (SPR): SPR is a technique that can measure the binding affinity and kinetics of protein-DNA interactions in real-time. It involves immobilizing the DNA on a surface and measuring the change in the refractive index caused by protein binding.
- Electrophoretic Mobility Shift Competition Assay (EMSCA): EMSCA is a variant of EMSA that involves the addition of a competitor DNA fragment that can bind to the protein of interest and compete with the labeled DNA probe for binding.
- Microscale Thermophoresis (MST): MST is a technique that can measure the binding affinity and kinetics of protein-DNA interactions in solution. It involves measuring the movement of fluorescently labeled DNA molecules in response to a temperature gradient generated by a laser.
These techniques can provide complementary information to EMSA and can be used to validate the EMSA results. The choice of technique depends on the specific research question and the availability of resources.
Learn more: