Author: Binod G C
For the initial detection of microbes, the innate immune system uses pattern recognition receptors (PRRs). Downstream signaling activated by PRRs leads to the induction of innate immune response producing inflammatory cytokines, interferons, and other mediators. (1). Different classes of PRRs in mammals include Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), Nod-like receptors (NLRs), AIM2-like receptors (ALRs), C-type lectin receptors (CLRs), and intracellular DNA sensors such as cGAS (2, 3). The first identified and best understood among them is TLRs.
TLR family comprises 10 members (TLR1-TLR10) in humans. TLRs recognize PAMPs such as lipid, lipoprotein, protein, and nucleic acid and localize to the surface of the cell or intracellular components such as endosome, Lysosome, ER, or endo-lysosome as a homodimer or heterodimer with a coreceptor. The ectodomain of TLR with leucine-rich repeats (LRRs) mediate PAMPs recognition while transmembrane domain and Toll/IL-1 receptor domain initiate the downstream signaling. (4). After PAMP or DAMP recognition, the TIL domain-containing adaptor proteins (MyD88 or TRIF) initiates downstream signal transduction which activates NF-κB, IRFs, or MAP kinases that regulate the expression of cytokines, chemokines and type 1 IFNs for the protection of the host against microbial infection. According to the recent studies, proper cellular localization of TLR plays an important role in a cell type-specific signaling regulation which subsequently determines the innate response.
TLRs recruit different sets of TIR domain-containing adaptors such as MyD88, TRIF, TIRAP/MAL, or TRAM. TLR signaling can be categorized into two types as MyD88 dependent TLR Signaling Pathway and TRIF dependent signaling pathway depending on the adaptor usage.
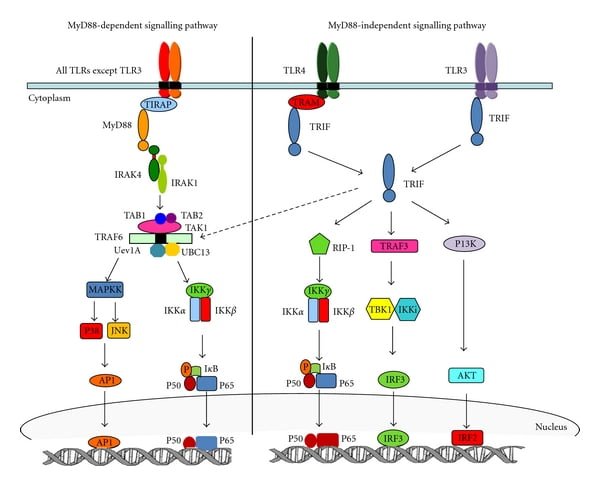
MyD88 dependent pathway
All the TLRs utilize MyD88 to activate MAPKs and NF-κB to induce inflammatory cytokine genes. TIRAP is the sorting adaptor that recruits MyD88 to the cell surface. Recent studies show that TIRAP is associated with both cell surface (TLR2andTLR4) and endosomal (TLR9) TLRs.
After being stimulated by ligands, MyD88 recruits IL1 receptor-associated kinase-4 (IRAK4) to TLRs through the interaction of the death domain of both the molecules. IRAK4 activates IRAK1 AND irak1 gets autophosphorylated and released form MyD88. The phosphorylated IRAK1 associated with the RING-domain E3 ubiquitin ligase TRAF6 along with other ubiquitin-conjugating enzymes UBC13 and UEV1A that subsequently promotes the K-63 linked polyubiquitination of TRAF6 which drives TAK1 activation.
A clear mechanism of TAK1 is still to be understood (5). Activated TAK1 leads to the activation of two different pathways IKK complex-NF-κB pathway and -MAPK pathway. IKK complex comprises of catalytic subunits IKKα and IKKβ and the regulatory subunit NEMO (also called IKKγ). Binding of TAK1 to IKK through ubiquitin chains causes phosphorylation and activates IKKβ. This IKK complex phosphorylates the NF-κB inhibitory protein IκBα, which undergoes proteasome degradation and causes nuclear translocation of NF-κB and induce proinflammatory gene expression. Along with this, TAK1 activation activates MAPK family members such as ERK1/2, p38, and JNK, which mediates activation of AP-1transcription factors or stabilizes mRNA to regulate inflammatory responses (2).
MyD88 independent/TRIF dependent pathway
The deletion of MyD88 causes a decrease in cytokine production due to aberrant signaling. Surprisingly, LPS stimulation in those MyD88 depleted cells still causes the activation of NF-κB and MAPKs pathways downstream although the kinetics is delayed. Interaction of TRIF with TRAP6 and TRAP3 instigates the TRIF dependent induction of inflammatory cytokines. TRAF6 recruits a kinase, RIP1 which interacts with TAK complex and activates NF-κB and MAPKs pathways downstream. The mechanism of TRAP3 is different from TRAF6, TRAF3 recruits TBK1 and IKKi kinases along with NEMO that leads to IRF3 phosphorylation. Consequently, IRF3 forms a dimer and translocate to the nucleus from the cytoplasm and induces the expression of type 1 IFN genes. Recently, it has been shown that IRF3 activation is regulated by inositol lipid, PtdIns5P which facilitates the complex formation of IRF3 and TBK1 (2).
Negative Regulation of TLR Signaling
As described above, the recognition of ligand and TLRs and the specificity of adaptor proteins leads to the activation of respective genes to produce proinflammatory cytokines and chemokines for the clearance and control of pathogens. The excessive production of cytokines and chemokines can be disastrous to the health of health even leading to death in some instances, which is called hyperimmune condition. The host, therefore, has evolved mechanisms to modulate TLR mediated immune response to negatively regulate TLR signaling (6).
IRAK-M appears to block the formation of IRAK-1/TRAF6 complex, but not the recruitment of IRAK-1 to MyD88, thus signaling of NF-κB and/or MAPKs cannot go ahead. Similarly, SOCS1-a member of SOCS family proteins also helps in the reduction of cytokine signaling through the JAK-STAT pathway. This occurs by polyubiquitination and subsequent proteasomal degradation of TIRAP. Furthermore, TOLLIP (Toll- interacting protein) interacts with TLR2 and TLR2 to clock the activation of the NF-κB pathway. These negative regulatory mechanisms utilizing IRAK-M / TOLLIP / SOCS1 maintains the immune cells in the dormant state and helps to end TLR signaling during inflammation and infection.
Recent Progress:
In a paper by Azam et al. 2019, they have assessed the polyphenols in the treatment of neurodegenerative diseases focusing on TLR regulation by polyphenols. Some polyphenols can intervene in TLR4/NF-κB/STAT signaling and suppress the overexpression of inflammatory mediators. TLRs are known to regulate signals to induce RCD (regulated cell death), which plays a role in the antitumor process. BCG being the first TLR targeted agent used to treat superficial bladder tumor, now after almost after half-century there has been substantial development in the understanding of TLR Signaling and cancer resulting in the emergence of various anti-cancer agents targeting TLR, however only four(BCG, monophosphoryl lipid A (MPLA), IMQ and REQ) has been approved by FDA (7).
Along with this, in a study by Refner et.al they suggest the role TLR2 pathway is associated with downstream chromatin modification by positively regulating the epithelial barrier function (8). Beyond canonical NF-κB pathway TLRs which helps in the regulation of cell-cell communication and signaling at synapse they also regulate various aspects of developmental biology. They regulate neuronal proliferation and differentiation in mammals and regulate adult hippocampal neurogenesis (9). TLR3 and TLR8 expression in the nervous system is dynamic during development, so both might function in the development of the nervous system. However, much remains unknown of these receptors in a developmental context. The existing literature can conclude that TLRs are one of the main elements in the innate immune system. TLR study includes the various aspects of TLR signaling including recognition of ligands, expression, adaptor proteins, and its implication in many diseases in recent years.
References:
- 1. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. DOI:10.1146/annurev.immunol.20.083001.084359
- 2. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi:10.1016/j.cell.2006.02.015
- 3. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54:289–96. doi:10.1016/j.molcel. 2014.03.040
- 4. Botos I, Segal DM, Davies DR. The structural biology of toll-like receptors. Structure (2011) 19:447–59. doi:10.1016/j.str.2011.02.004
- 5. Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li S, et al. Sequential autophosphorylation steps in the interleukin-1receptor-associated kinase-1regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem (2004) 279:5227–36. doi:10.1074/jbc.M309251200
- 6. Liew FY, Xu D, Brint EK, O’Neill LA. 2005. Negative regulation of Toll-like receptor-mediated immune responses. Nat.Rev.Immunol. 5:446–58
- 7. Azam S, Jakaria M, Kim I-S, Kim J, Haque ME, and Choi D-K (2019) Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 10:1000. doi: 10.3389/fimmu.2019.01000
- 8. Ruffner, Melanie A., et al. “Toll‐like receptor 2 stimulation augments esophageal barrier integrity.” Allergy 74.12 (2019): 2449-2460.
- 9. Okun, E., Griffioen, K. J. and Mattson, M. P.(2011). Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 34, 269-281. doi:10.1016/j.tins.2011.02.005