What is Cellular Senescence?
Senescence is the natural process of aging in living organisms. In biology, it refers to cell aging, where cells permanently stop dividing but do not die. Cellular senescence is characterized by stable growth arrest and various phenotypic changes, including the secretion of proinflammatory substances. It involves the gradual deterioration of functional characteristics and morphological features, leading to a loss of cell reproduction ability. Senescence occurs as organisms age and experience a decline in function.
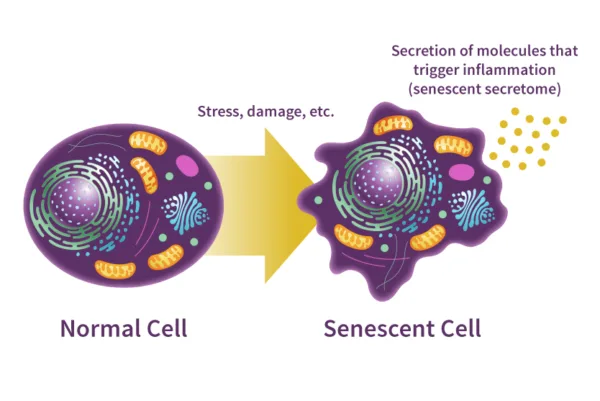
Mechanisms Behind Cellular Senescence
Persistent DNA damage and other stress-induced signals can induce a state of permanent cell proliferation arrest, which is known as cellular senescence.
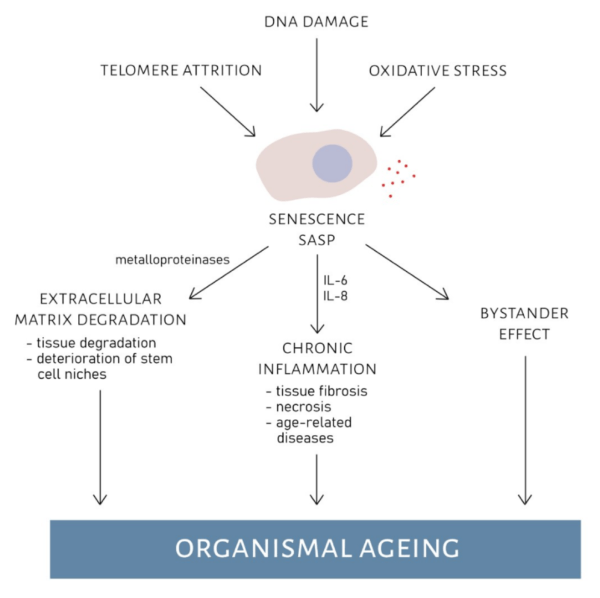
This complex biological process can be triggered by various factors, such as DNA damage, telomere shortening, oxidative stress, and oncogene activation. The mechanisms underlying cellular senescence are interconnected pathways and molecular events. The key mechanisms involved are discussed below:
- Telomere Shortening
- DNA Damage Response
- Activation of Tumor Suppressor Pathways
- Senescence-Associated Secretory Phenotype (SASP)
- Epigenetic Alterations
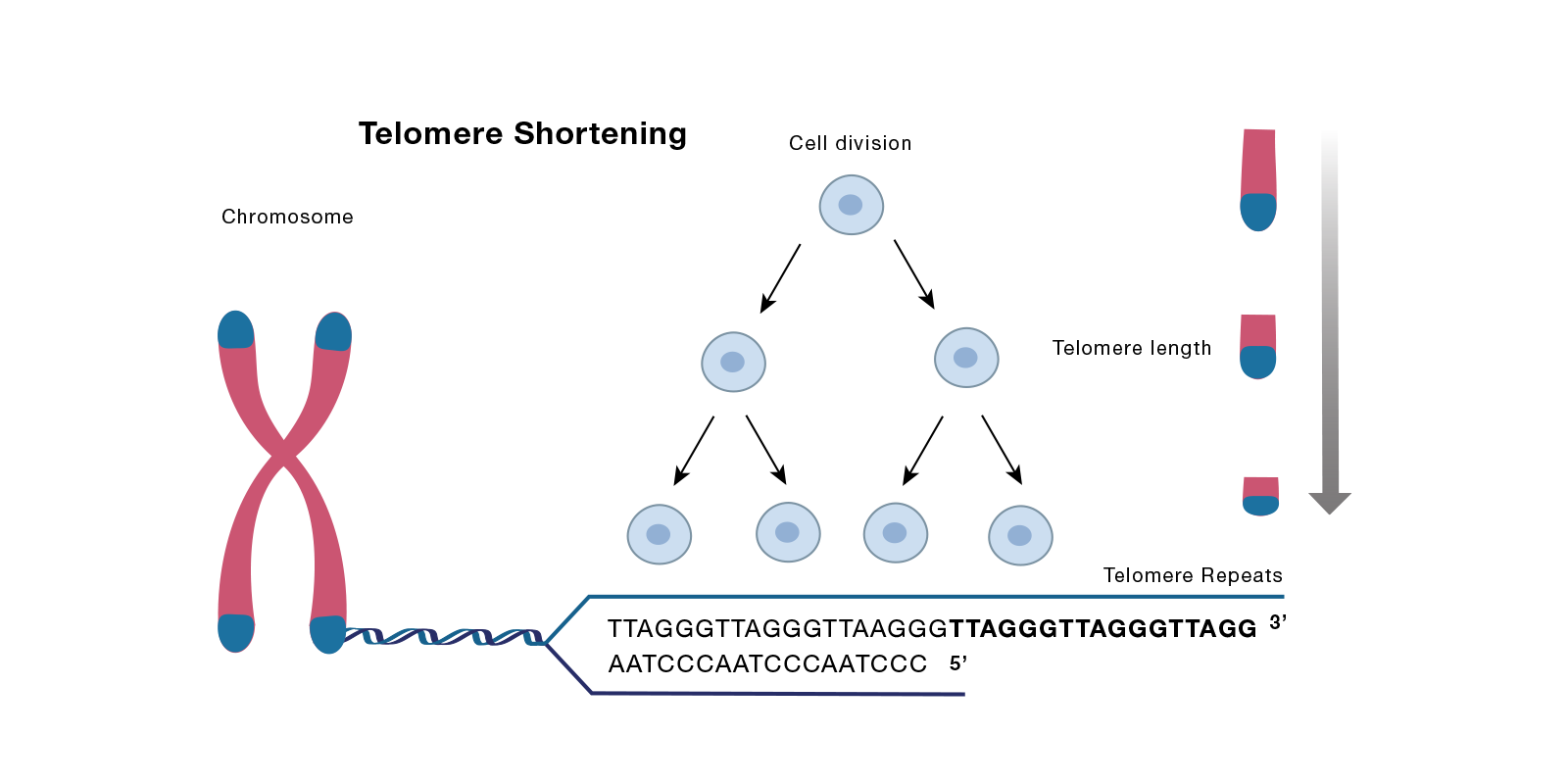
1. Telomere Shortening
Telomeres, found at the ends of linear chromosomes, act as protective caps, safeguarding against genome instability. Composed of repetitive DNA sequences called tandem TTAGGG repeats, they are associated with a protein complex called “Shelterin.” This complex, crucial for telomere function, stabilizes a distinct structure known as the telomere-loop or t-loop.
The telomere-loop, resembling a lariat, forms at the chromosome end, providing shielding and protection. By interacting with telomeric DNA, the Shelterin complex ensures the integrity of telomeres, preventing their identification as DNA damage sites.
Telomeres, the protective caps at the ends of chromosomes, undergo a process of shortening during DNA replication. This shortening occurs because DNA polymerase faces limitations when replicating the single-stranded 3′ ends of chromosomes. This phenomenon was proposed by Olovnikov in 1971 and later confirmed by experiments conducted by Blackburn. Telomere shortening acts as a replication “timer,” even though telomerase, an enzyme involved in telomere elongation, exists.
When telomeres reach a critical point of shortening, typically around 12.8 repeats, they become dysfunctional, which can lead to chromosome fusion and instability in the genome. This triggers protective mechanisms within cells, such as cell cycle arrest, cellular senescence (a state of irreversible growth arrest), and apoptosis (programmed cell death). These protective measures aim to prevent the detrimental effects of telomere dysfunction.
However, it is possible for cells to bypass cell cycle arrest and become cancerous through the activation of oncogenes or telomerase, which can maintain telomere length. Additionally, even with long telomeres, the speed and intensity of telomere utilization and loss can influence the occurrence of cell cycle arrest. Furthermore, telomeres can also undergo shortening due to DNA double-strand break damage, which triggers a cellular response similar to that of DNA double-strand breaks. This response involves the phosphorylation of H2AX, a protein that plays a role in DNA damage signaling.
The process of cell cycle arrest is a universal protective mechanism that helps safeguard against malignancy and serves as a natural endpoint of cell development, commonly associated with aging. This connection between telomere dynamics, cell cycle arrest, and aging aligns with various aging theories and their shared understanding of disease pathogenesis.
Telomere Theory
The Telomere theory of aging rests on three key principles, often referred to as the pillars of the theory. These principles are as follows:
- Aging is programmed: The Telomere theory proposes that aging is not a random process but a programmed biological phenomenon. It suggests that specific genetic programs regulate the intrinsic aging mechanism of our cells.
- Telomere shortening triggers irreversible cell cycle arrest: According to the theory, as cells divide, the protective caps called telomeres at the ends of chromosomes progressively shorten. When telomeres become critically short, cells experience irreversible cell cycle arrest, meaning they can no longer divide and replicate.
- Hayflick limit restricts cell division: The Telomere theory introduces the concept of the Hayflick limit, which states that the total number of cell divisions without sufficient telomerase activity is limited. Telomerase is an enzyme that replenishes telomeres, enabling cells to surpass the Hayflick limit. However, most somatic cells in the human body lack adequate telomerase activity, imposing a finite limit on their division potential.
These three principles form the foundation of the Telomere theory, suggesting that the programmed shortening of telomeres leads to irreversible cell cycle arrest and contributes to the aging process.
2. DNA Damage Response
The DNA damage response (DDR) plays a vital role in cellular senescence. When cells experience DNA damage, such as double-strand breaks or genetic lesions, they activate the DDR, a complex signaling network.
During cellular senescence, the DDR is triggered to protect against DNA damage propagation and maintain genomic stability. Activation of DDR pathways, including ATM and ATR checkpoint proteins, initiates a cascade of events.
These events include cell cycle arrest, DNA repair, and the induction of cellular senescence. Cell cycle arrest allows time for DNA repair, and if the damage is severe or unrepairable, cellular senescence occurs. Senescent cells undergo irreversible growth arrest and exhibit distinct changes, like an enlarged and flattened shape.
The DDR in cellular senescence leads to various molecular and cellular changes, including the activation of the senescence-associated secretory phenotype (SASP). SASP involves the secretion of pro-inflammatory cytokines, growth factors, and matrix remodeling enzymes.
In summary, the DNA damage response is a crucial component of cellular senescence, safeguarding against DNA damage propagation and contributing to the development of a senescent phenotype.
3. Activation of Tumor Suppressor Pathways
Tumor suppressor pathways activate to regulate cellular senescence, with the p53 and Rb pathways being the major ones involved.
p53 Pathway
The p53 pathway acts as a transcription factor, stabilizing and activating in response to DNA damage signals. It induces downstream target genes involved in cell cycle arrest, DNA repair, and apoptosis. In cellular senescence, p53 increases p21 expression, leading to growth arrest and senescence.
Rb pathway
The Rb pathway functions as a negative regulator of the cell cycle, inhibiting E2F transcription factors that are necessary for cell cycle progression. Senescence induces p16INK4a, which binds and inhibits cyclin-dependent kinases that phosphorylate and inactivate Rb. The resulting Rb inactivation allows the release of E2F, promoting cell cycle progression. However, increased p16INK4a levels during senescence prevent Rb phosphorylation, leading to cell cycle arrest and senescence.
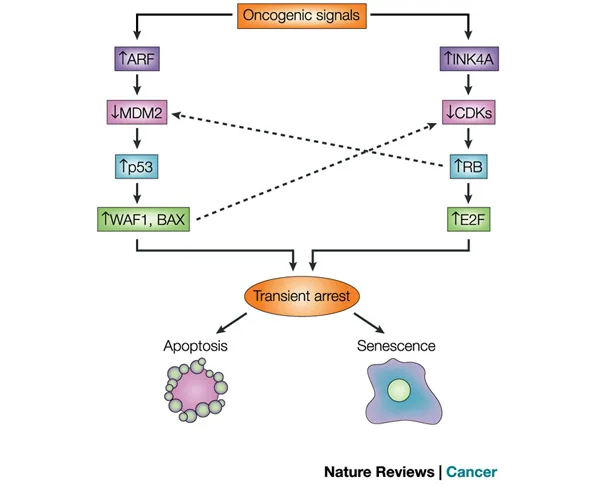
Other tumor suppressors, such as PTEN and BRCA1, are also involved in cellular senescence, regulating cell survival, growth, proliferation, and DNA repair. Dysregulation of these pathways can cause cells to escape senescence and contribute to tumorigenesis. Overall, tumor suppressor pathway activation, especially the p53 and Rb pathways, initiates and maintains cellular senescence, preventing damaged cell propagation and acting as a critical defense against cancer development.
4. Senescence-Associated Secretory Phenotype (SASP)
Cells can enter cellular senescence, a state of permanent growth arrest, in response to DNA damage and to prevent cancer. This process relies on specific tumor suppressor pathways, like p16(INK4a)/pRB and p53/p21(WAF1), triggered by various stress-inducing stimuli such as telomere dysfunction, oncogenic mutations, and genotoxic stress. Senescent cells release a unique set of molecules called the senescence-associated secretory phenotype (SASP), including cytokines, chemokines, growth factors, proteases, and extracellular vesicles. Accumulation of senescent cells and the SASP with age contributes to age-related disorders, accelerated by factors like ultraviolet radiation (UVR) causing tissue dysfunction.
The SASP not only causes cell cycle arrest but also influences the tissue microenvironment, reinforcing cellular senescence within senescent cells (autocrine) and inducing senescence in surrounding cells (paracrine). Senescent cells release chemokines affecting immune cells like natural killer cells and macrophages, aiding in the clearance of senescent cells.
Furthermore, the SASP recruits immune cells to damaged tissues, promoting progenitor cell proliferation for tissue repair. For instance, in skin repair, SASP factors from senescent fibroblasts recruit immune cells to remove damaged tissues and stimulate skin progenitor cell proliferation. In liver injury, senescent hepatic stellate cells produce SASP factors, recruiting immune cells to eliminate senescent cells and prevent fibrosis.
Persistent SASP can have detrimental effects, including chronic inflammation and cancer progression. Cancer-associated fibroblasts (CAFs) exhibit SASP and contribute to tumor promotion. SASP factors from senescent hepatic stellate cells in the obesity-associated liver tumor microenvironment are linked to tumor progression, chronic inflammation, and negative outcomes.
The induction of SASP involves the cGAS-STING pathway associated with innate immunity. Persistent DNA damage in senescent cells downregulates the histone dimethylating enzyme G9a, leading to the accumulation of cytoplasmic DNA fragments. These fragments activate the cGAS-STING pathway, triggering cytokine production and initiating inflammatory responses. Abnormal accumulation of cytoplasmic DNA fragments is facilitated by downregulated DNases. This intrinsic trigger through the cGAS-STING pathway plays a pivotal role in promoting SASP and its effects.
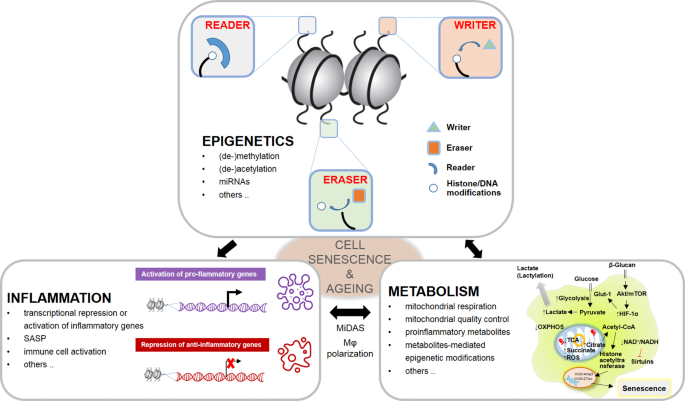
5. Epigenetic Alterations
Eukaryotic chromosomes possess two separate sections known as heterochromatin and euchromatin. Heterochromatin refers to densely packed chromatin that restricts gene expression, whereas euchromatin enables active transcription. Changes in heterochromatin have been noted during the process of aging and cellular senescence, resulting in modifications to gene expression patterns. We delves into the epigenetic modifications and alterations in chromatin organization linked to cellular senescence in these points.
- Aging and Heterochromatin: Aging triggers heterochromatin loss and instability in various organisms, including humans, C. elegans, and S. cerevisiae. Persistent DNA damage, reduced production of histone chaperone proteins, and decreased histone biosynthesis drive this heterochromatin decline. Furthermore, senescent cells release chromatin fragments, further contributing to the loss of heterochromatin.
- Histone Epigenetic Modifications: Histones undergo crucial post-translational modifications that impact gene expression and chromatin structure. Histone acetylation and deacetylation, facilitated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively, regulate chromatin accessibility to transcription factors. Methylation of histone lysine and arginine residues also influences gene transcription. Dysregulation of these modifications disrupts cellular senescence.
- Senescence-Associated Heterochromatic Foci (SAHF): Senescent cells exhibit distinctive nuclear structures called SAHF, characterized by condensed heterochromatin enriched in hypoacetylated histones, H3K9me3, H3K27me3, HP1 proteins, and macroH2A histone variant. SAHF suppress the transcription of proliferation-related genes, contributing to the cessation of cell division. Moreover, SAHF protect senescent cells from apoptosis by modulating the DNA damage response. Formation of SAHF involves chromatin organization changes, including loss of lamin B1 and redistribution of heterochromatin.
- Senescence-Associated Distention of Satellites (SADS): In addition to SAHF, senescent cells experience alterations in constitutive heterochromatin regions, specifically pericentric satellite DNA. Known as SADS, these changes involve significant decondensation of pericentric satellite DNA and have been linked to cellular senescence and tissue aging. Although the precise role of SADS remains unclear, its formation is consistent across different senescent cell types and stimuli.
- Epigenetic Influences on Cellular Senescence: Epigenetic regulators and effectors play crucial roles in establishing and maintaining cellular senescence. High mobility group (HMG) proteins like HMGA1 and HMGB2 cooperate with SAHF to support the senescence phenotype. HMGA proteins aid in maintaining proliferative arrest, while HMGB proteins regulate SASP gene expression. Additionally, other epigenetic effectors, including super-enhancers, histone variants, sirtuins, and methyltransferases, modulate the SASP and pro-inflammatory signaling pathways.
Conclusion
To summarize, cellular senescence is a complicated mechanism that prevents cellular aging and maintains tissue equilibrium. Telomere shortening functions as a replication timer, prompting protective responses when telomeres approach a critical degree of malfunction. These include cell cycle arrest, cellular senescence, and apoptosis, all of which help to avoid genetic instability and cancer.
Telomere dynamics do not promote cellular senescence on their own. DNA damage and gene activation are additional factors that impact the beginning of senescence. Although universal cell cycle arrest protects against cancer, overcoming it via oncogene activation or telomerase activity can result in malignant transformation.
Understanding the mechanics of cellular senescence is critical for understanding aging and disease processes. The study of epigenetic changes, chromatin architecture, and telomere dynamics in senescence gives insight into molecular causes and possible treatment targets for age-related illnesses.